본 글은 'NGS | Clinical Data Analysis Overview (1/2)'에 이어 작성된 글입니다. 먼저 해당 글을 읽고 오시는 것을 권장합니다.
Intro
암 환자의 DNA 시퀀싱 이후 분석과정을 살펴보겠습니다.
다시 한 번 암 환자의 DNA 분석의 목표를 생각해보면, 개인의 고유한 특성이 담긴 유전체 서열을 시퀀싱하고 참조 유전체와 비교하여 발견되는 variant들이 질병과 어떻게 연관되어 있는지, 그리고 관련된 치료제는 어떤 것들이 있는지 확인하는 것입니다. 지난 글에 이어 본 글에서는 시퀀싱 이후 데이터 분석 과정을 살펴보겠습니다.
다시 한 번 암 환자의 DNA 분석의 목표를 생각해보면, 개인의 고유한 특성이 담긴 유전체 서열을 시퀀싱하고 참조 유전체와 비교하여 발견되는 variant들이 질병과 어떻게 연관되어 있는지, 그리고 관련된 치료제는 어떤 것들이 있는지 확인하는 것입니다. 지난 글에 이어 본 글에서는 시퀀싱 이후 데이터 분석 과정을 살펴보겠습니다.
Demultiplexing
샘플별로 데이터를 분류하는 동시에 'bcl' 파일을 'fastq' 형태로 변환하는 단계입니다.
라이브러리 제작시 fragment 양 쪽 말단에 어댑터를 추가했습니다. 어댑터에는 샘플 고유의 tag 작용을 하는 index 서열이 포함되어 있습니다. 여러 개 샘플 라이브러리들을 하나의 flow cell에 로딩하여 시퀀싱 합니다. 이렇게 두 개 이상의 샘플을 동시에 시퀀싱하는 것을 'multiplexing'이라고 부릅니다.
라이브러리 제작시 fragment 양 쪽 말단에 어댑터를 추가했습니다. 어댑터에는 샘플 고유의 tag 작용을 하는 index 서열이 포함되어 있습니다. 여러 개 샘플 라이브러리들을 하나의 flow cell에 로딩하여 시퀀싱 합니다. 이렇게 두 개 이상의 샘플을 동시에 시퀀싱하는 것을 'multiplexing'이라고 부릅니다.
왜 굳이 번거롭게 index 서열을 사용하면서까지 여러 개 샘플을 동시에 시퀀싱 할까요? 바로 경제적인 이유 때문입니다. NextSeq 300 cycle 기준 한 번 시퀀싱 하는데 들어가는 시약 비용은 최대 4,820$에 달합니다. NovaSeq 300 cycle 기준 최대 30,780$가 들어갑니다. (2020년 기준) 결코 무시할 수 없는 적지않은 비용입니다. 따라서 시퀀서별 생산할 수 있는 데이터량에 맞춰 샘플별 목표 데이터량을 조합하고 여러 개의 샘플을 multiplexing 합니다. 이렇게 샘플의 라이브러리가 섞인 상태에서 시퀀싱을 진행합니다. 데이터를 분석하기 위해서 index 서열 기준으로 샘플별 데이터를 분류합니다. 동시에 바이너리 형태의 'bcl' 파일을 'fastq' 형태로 변환합니다. 'Multiplexing'의 반대 과정이라는 의미에서 'demultiplexing'이라고 부릅니다. 'bcl' 파일과 'fastq' 파일은 가공하지 않은 원 데이터라는 의미에서 'raw data'로 부릅니다. 블로그에서는 별다른 언급이 없는 한 fastq 파일을 raw data로 지칭하겠습니다.

Demultiplexing은 Illumina에서 제공하는 'bcl2fastq' 프로그램을 사용합니다. Input은 시퀀서에서 생산된 '런 폴더'와 샘플 정보가 들어있는 'sample sheet'가 필요합니다.
Demultiplexing은 Illumina에서 제공하는 'bcl2fastq' 프로그램을 사용합니다. Input은 시퀀서에서 생산된 '런 폴더'와 샘플 정보가 들어있는 'sample sheet'가 필요합니다.
Data QC
Raw data의 퀄리티를 체크하는 단계입니다.
컴퓨터 과학 분야에서 사용되는 문구입니다. 쓸모없는 데이터를 집어넣고 실행하면 쓸모없는 결과를 얻는다는 의미입니다. 곰곰히 생각해보면 세상 거의 대부분의 현상에 적용되는 것 같습니다. 아무리 요리사의 실력이 뛰어나고 훌륭한 조리도구를 사용하더라도 질 낮은 식재료를 사용한다면 과연 음식이 맛있을까요? 겉으로 보기에 튼튼해 보이는 건물도 부실한 원자재를 사용했다면 머지않아 각종 문제가 발생하고 결국 무너질 것입니다. 사람의 인생도 마찬가지입니다. 아무리 화려하게 포장해 놓았더라도 준비와 노력없이 낭비한 시간은 결국 드러나게 되어 있습니다. (물론 모든 사람은 그 존재 자체만으로 가치있으며 존중받아 마땅합니다. ^^) 임상 데이터를 다루는 분야에서 raw data의 퀄리티 체크는 간과할 수 없는 매우 중요한 단계입니다. 특히 잘못된 리포트 결과가 환자의 치료에 영향을 미칠 수 있기 때문에 언제나 책임감을 가지고 분석에 신중을 기해야 합니다.
Raw data 퀄리티 체크는 'fastqc' 프로그램을 사용합니다. Input은 PE read1, 2가 필요합니다. Output은 read1, 2 각각 결과파일로 출력합니다.

'fastqc'는 v0.11.9 기준 raw data 퀄리티와 관련된 11가지 항목을 분석한 뒤 보여줍니다. 중점적으로 살펴보는 항목은 'Per base sequence quality', 'Per tile sequence quality' 입니다.
'Per base sequence quality'는 모든 시퀀싱 cycle에 대해서 read가 가지고 있는 phred score를 box plot 형태로 나타낸 결과입니다. 결과를 해석하기 위해서 phred score에 대한 이해가 필요합니다. 앞서 암 환자에게서 DNA를 채취하고 시퀀서가 서열을 읽어 raw data까지 생산 했습니다. 이 때 Illumina 기기는 SBS 기술로 서열을 읽는데 cycle별 방출되는 형광물질 색상과 시그널 강도를 A, T, C, G로 해석했습니다. 모든 염기서열을 정확히 읽어내면 좋겠지만 클러스터가 너무 근접해 있다던가 서열의 diversity가 떨어지면 잘못된 서열로 해석할 가능성이 높아집니다. 이처럼 시퀀싱 서열이 잘못 해석되었을 확률(P)을 상용로그 단위로 나타낸 값을 base quality(Q)라고 부르며 다음과 같이 계산합니다.

즉, Q10은 10번 base calling 했을 때 1번 에러가 발생하며 9번 정확하게 calling한다는 의미입니다.

절대적 기준이 존재하지 않지만 저는 모든 서열이 최소 Q20 이상 갖도록 하며 동시에 Q30 이상인 서열이 전체 서열 중 80% 이상 되도록 조절합니다. 모두 최상의 퀄리티를 유지하도록 조절하면 좋겠지만 너무 많은 read를 잘라내거나 버려야 할 수 있으므로 적절한 조절이 필요합니다.
Rubbish in, Rubbish out (RIRO)
컴퓨터 과학 분야에서 사용되는 문구입니다. 쓸모없는 데이터를 집어넣고 실행하면 쓸모없는 결과를 얻는다는 의미입니다. 곰곰히 생각해보면 세상 거의 대부분의 현상에 적용되는 것 같습니다. 아무리 요리사의 실력이 뛰어나고 훌륭한 조리도구를 사용하더라도 질 낮은 식재료를 사용한다면 과연 음식이 맛있을까요? 겉으로 보기에 튼튼해 보이는 건물도 부실한 원자재를 사용했다면 머지않아 각종 문제가 발생하고 결국 무너질 것입니다. 사람의 인생도 마찬가지입니다. 아무리 화려하게 포장해 놓았더라도 준비와 노력없이 낭비한 시간은 결국 드러나게 되어 있습니다. (물론 모든 사람은 그 존재 자체만으로 가치있으며 존중받아 마땅합니다. ^^) 임상 데이터를 다루는 분야에서 raw data의 퀄리티 체크는 간과할 수 없는 매우 중요한 단계입니다. 특히 잘못된 리포트 결과가 환자의 치료에 영향을 미칠 수 있기 때문에 언제나 책임감을 가지고 분석에 신중을 기해야 합니다.
Raw data 퀄리티 체크는 'fastqc' 프로그램을 사용합니다. Input은 PE read1, 2가 필요합니다. Output은 read1, 2 각각 결과파일로 출력합니다.
'fastqc'는 v0.11.9 기준 raw data 퀄리티와 관련된 11가지 항목을 분석한 뒤 보여줍니다. 중점적으로 살펴보는 항목은 'Per base sequence quality', 'Per tile sequence quality' 입니다.
'Per base sequence quality'는 모든 시퀀싱 cycle에 대해서 read가 가지고 있는 phred score를 box plot 형태로 나타낸 결과입니다. 결과를 해석하기 위해서 phred score에 대한 이해가 필요합니다. 앞서 암 환자에게서 DNA를 채취하고 시퀀서가 서열을 읽어 raw data까지 생산 했습니다. 이 때 Illumina 기기는 SBS 기술로 서열을 읽는데 cycle별 방출되는 형광물질 색상과 시그널 강도를 A, T, C, G로 해석했습니다. 모든 염기서열을 정확히 읽어내면 좋겠지만 클러스터가 너무 근접해 있다던가 서열의 diversity가 떨어지면 잘못된 서열로 해석할 가능성이 높아집니다. 이처럼 시퀀싱 서열이 잘못 해석되었을 확률(P)을 상용로그 단위로 나타낸 값을 base quality(Q)라고 부르며 다음과 같이 계산합니다.
즉, Q10은 10번 base calling 했을 때 1번 에러가 발생하며 9번 정확하게 calling한다는 의미입니다.
절대적 기준이 존재하지 않지만 저는 모든 서열이 최소 Q20 이상 갖도록 하며 동시에 Q30 이상인 서열이 전체 서열 중 80% 이상 되도록 조절합니다. 모두 최상의 퀄리티를 유지하도록 조절하면 좋겠지만 너무 많은 read를 잘라내거나 버려야 할 수 있으므로 적절한 조절이 필요합니다.
Quality & Adapter Trimming
Quality가 떨어지는 서열과 adapter 서열을 잘라내는 단계입니다.
Adapter는 라이브러리 제작시 fragment 양 쪽 말단에 붙여준 서열입니다. 라이브러리 제작 키트마다 고유한 서열을 가지고 있으므로 키트 제조사에서 제공하는 adpater 서열을 확인한 뒤 제거합니다. 저는 Illumina의 TruSeq 키트를 사용하고 있으며 다음 페이지에서 관련 자료를 확인할 수 있습니다.
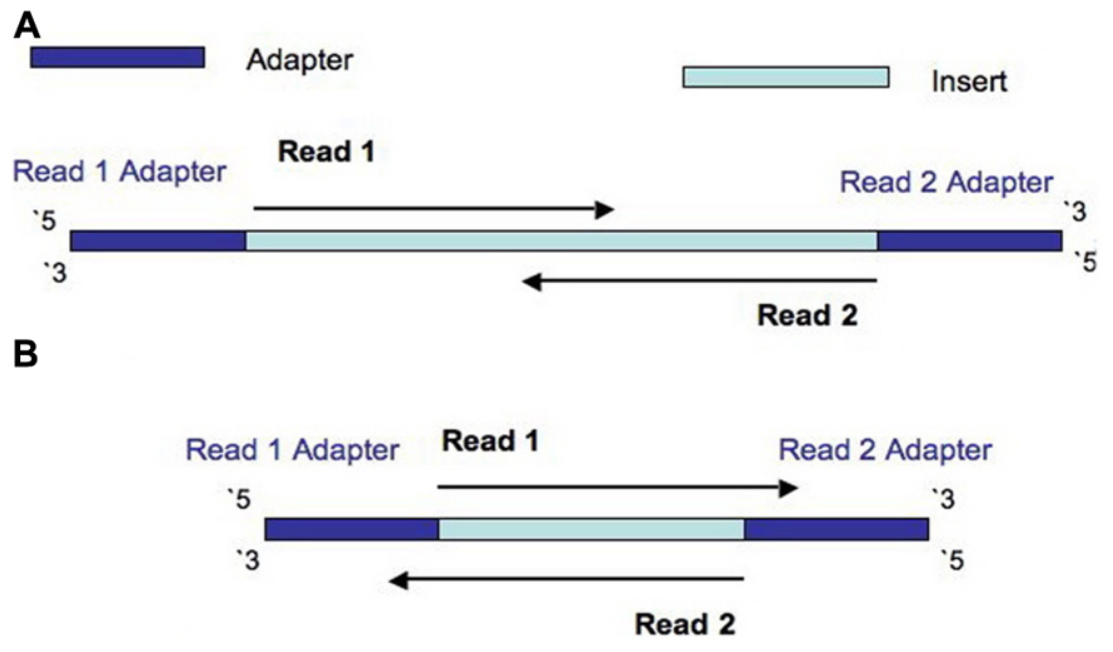
A와 같이 보통 시퀀싱 길이보다 길이가 긴 fragment를 사용하므로 adapter 서열은 시퀀싱되지 않는 것이 이상적인 상황입니다. 하지만 B와 같이 시퀀싱 길이보다 짧은 fragment도 시퀀싱에 포함되므로 adapter 서열까지 시퀀싱 된 read가 일부 존재합니다. 어차피 레퍼런스 서열에 mapping할 때 걸러내지므로 제거할 필요가 없다는 의견도 일부 존재하지만 개인적으로 정확한 분석을 위해서 깨끗하게 잘라내는 것이 좋다고 생각합니다.
Base quality가 떨어지는 서열도 정확한 분석을 위해 잘라냅니다. Read1과 read2 모두 마지막 cycle로 갈수록 base quality가 떨어집니다. 시퀀싱 시약의 한계상 점차 시그널 강도가 약해지므로 base calling의 정확도가 감소하고 base quality가 떨어지는 것입니다. 이와 관련된 현상으로 Illumina의 2-color channel 시스템을 사용하는 NextSeq, NovaSeq, iSeq 기기들은 일부 read에서 poly-G 서열이 나타납니다. 2-color channel 시스템은 형광물질 R(red)과 G(Green) 두 가지를 사용하는데, R은 C, G는 T, Y(Yellow = ref + green)는 A, B(Black = no R + no G)는 G로 해석합니다. 즉, 시그널이 없는 상태를 G로 해석하기 때문에 시그널 강도가 약해지는 마지막 cycle로 갈수록 모두 G로 잘못 해석해 버리는 현상이 발생합니다.
Adapter 서열 제거는 'fastp', 'cutadapt', 'trimmomatic' 등 여러 가지 프로그램이 있습니다. 저는 fastp를 사용하는데 성능이 나은 편이고 adapter와 quality trimming을 모두 사용할 수 있으며 poly-G 서열을 제거하는 기능까지 가지고 있기 때문입니다.
Adapter는 라이브러리 제작시 fragment 양 쪽 말단에 붙여준 서열입니다. 라이브러리 제작 키트마다 고유한 서열을 가지고 있으므로 키트 제조사에서 제공하는 adpater 서열을 확인한 뒤 제거합니다. 저는 Illumina의 TruSeq 키트를 사용하고 있으며 다음 페이지에서 관련 자료를 확인할 수 있습니다.
A와 같이 보통 시퀀싱 길이보다 길이가 긴 fragment를 사용하므로 adapter 서열은 시퀀싱되지 않는 것이 이상적인 상황입니다. 하지만 B와 같이 시퀀싱 길이보다 짧은 fragment도 시퀀싱에 포함되므로 adapter 서열까지 시퀀싱 된 read가 일부 존재합니다. 어차피 레퍼런스 서열에 mapping할 때 걸러내지므로 제거할 필요가 없다는 의견도 일부 존재하지만 개인적으로 정확한 분석을 위해서 깨끗하게 잘라내는 것이 좋다고 생각합니다.
Base quality가 떨어지는 서열도 정확한 분석을 위해 잘라냅니다. Read1과 read2 모두 마지막 cycle로 갈수록 base quality가 떨어집니다. 시퀀싱 시약의 한계상 점차 시그널 강도가 약해지므로 base calling의 정확도가 감소하고 base quality가 떨어지는 것입니다. 이와 관련된 현상으로 Illumina의 2-color channel 시스템을 사용하는 NextSeq, NovaSeq, iSeq 기기들은 일부 read에서 poly-G 서열이 나타납니다. 2-color channel 시스템은 형광물질 R(red)과 G(Green) 두 가지를 사용하는데, R은 C, G는 T, Y(Yellow = ref + green)는 A, B(Black = no R + no G)는 G로 해석합니다. 즉, 시그널이 없는 상태를 G로 해석하기 때문에 시그널 강도가 약해지는 마지막 cycle로 갈수록 모두 G로 잘못 해석해 버리는 현상이 발생합니다.
Adapter 서열 제거는 'fastp', 'cutadapt', 'trimmomatic' 등 여러 가지 프로그램이 있습니다. 저는 fastp를 사용하는데 성능이 나은 편이고 adapter와 quality trimming을 모두 사용할 수 있으며 poly-G 서열을 제거하는 기능까지 가지고 있기 때문입니다.
Read Mapping
시퀀싱 read를 표준 유전체 서열에 붙이는 단계입니다.
분석 목적을 다시 상기해 봅시다. 암 환자에게서 고유한 유전체 정보를 가진 DNA를 채취하여 시퀀싱한 뒤 서열 정보를 얻어냈습니다. 이 서열 정보를 사람의 표준 유전체 서열과 비교하여 다른 부분을 찾아냅니다. 그리고 이 차이가 어떤 질병과 연관되어 있는지, 관련된 치료제는 어떤 것이 있는지 알아내기 위함입니다.
GRC Genome Reference Consortium는 사람(human), 쥐(mouse), 제브라피쉬(zebrafish), 닭(chicken)에 대한 표준 유전체 서열을 연구하고 발표합니다. 사람의 표준 유전체 서열은 2009년 2월에 발표한 GRCh37에 이어 2019년 3월에 발표한 GRCh38.p13(GRCh38의 열 세 번째 패치버전)이 있습니다. 이 외에도 UCSC The University of California at Santa Cruz에서 GRCh37을 바탕으로 만든 hg19, Broad Institute에서 GRCh37을 바탕으로 만든 b37 등이 있습니다. 모두 사람의 22개 염색체 및 X 염색체 서열은 동일하며 Y 염색체와 그 외 contig에서 차이를 보입니다. 최근 발표되는 일부 분석 프로그램은 GRCh38을 사용하기도 하나 이제까지 hg19로 진행된 연구가 다수이고 아직도 많이 사용하고 있으므로 저는 사람의 표준 유전체 서열로 hg19를 사용합니다.
Read mapping은 DNA 데이터인 경우 'bwa', 'bowtie' 등의 프로그램이 있으며 RNA 데이터인 경우 'TopHat', 'STAR', 'HISAT2' 등의 프로그램이 있습니다. 주로 DNA 시료를 사용하며 NextSeq 150bp PE 데이터를 사용하기에 보통 bwa를 사용합니다. bwa는 mem, sw, backtrack 세 가지 알고리즘을 가지고 있는데, 가장 최신이자 좋은 퍼포먼스를 보이고 데이터에 알맞는 mem을 사용합니다.
Mapping이 완료되면 SAM sequence alignment map 파일이 생성됩니다. Read가 표준 유전체 서열의 어느 위치에 mapping되었는지, 정확도는 얼마인지 등의 정보를 담고 있습니다.
분석 목적을 다시 상기해 봅시다. 암 환자에게서 고유한 유전체 정보를 가진 DNA를 채취하여 시퀀싱한 뒤 서열 정보를 얻어냈습니다. 이 서열 정보를 사람의 표준 유전체 서열과 비교하여 다른 부분을 찾아냅니다. 그리고 이 차이가 어떤 질병과 연관되어 있는지, 관련된 치료제는 어떤 것이 있는지 알아내기 위함입니다.
GRC Genome Reference Consortium는 사람(human), 쥐(mouse), 제브라피쉬(zebrafish), 닭(chicken)에 대한 표준 유전체 서열을 연구하고 발표합니다. 사람의 표준 유전체 서열은 2009년 2월에 발표한 GRCh37에 이어 2019년 3월에 발표한 GRCh38.p13(GRCh38의 열 세 번째 패치버전)이 있습니다. 이 외에도 UCSC The University of California at Santa Cruz에서 GRCh37을 바탕으로 만든 hg19, Broad Institute에서 GRCh37을 바탕으로 만든 b37 등이 있습니다. 모두 사람의 22개 염색체 및 X 염색체 서열은 동일하며 Y 염색체와 그 외 contig에서 차이를 보입니다. 최근 발표되는 일부 분석 프로그램은 GRCh38을 사용하기도 하나 이제까지 hg19로 진행된 연구가 다수이고 아직도 많이 사용하고 있으므로 저는 사람의 표준 유전체 서열로 hg19를 사용합니다.
Read mapping은 DNA 데이터인 경우 'bwa', 'bowtie' 등의 프로그램이 있으며 RNA 데이터인 경우 'TopHat', 'STAR', 'HISAT2' 등의 프로그램이 있습니다. 주로 DNA 시료를 사용하며 NextSeq 150bp PE 데이터를 사용하기에 보통 bwa를 사용합니다. bwa는 mem, sw, backtrack 세 가지 알고리즘을 가지고 있는데, 가장 최신이자 좋은 퍼포먼스를 보이고 데이터에 알맞는 mem을 사용합니다.
Mapping이 완료되면 SAM sequence alignment map 파일이 생성됩니다. Read가 표준 유전체 서열의 어느 위치에 mapping되었는지, 정확도는 얼마인지 등의 정보를 담고 있습니다.

각 컬럼이 의미하는 내용은 아래와 같습니다.
|
Col |
Field |
Type |
Regexp/Range |
Brief description |
|
1 |
QNAME |
String |
[!-?A-~]{1,254} |
Query template NAME |
|
2 |
FLAG |
Int |
[0, 216 − 1] |
bitwise FLAG |
|
3 |
RNAME |
String |
\*|[:rname:∧ *=][:rname:]* |
Reference sequence NAME |
|
4 |
POS |
Int |
[0, 231 − 1] |
1-based leftmost mapping POSition |
|
5 |
MAPQ |
Int |
[0, 28 − 1] |
MAPping Quality |
|
6 |
CIGAR |
String |
\*|([0-9]+[MIDNSHPX=])+ |
CIGAR string |
|
7 |
RNEXT |
String |
\*|=|[:rname:∧ *=][:rname:]* |
Reference name of
the mate/next read |
|
8 |
PNEXT |
Int |
[0, 231 − 1] |
Position of the mate/next read |
|
9 |
TLEN |
Int |
[−231 + 1, 231
− 1 |
observed Template LENgth |
|
10 |
SEQ |
String |
\*|[A-Za-z=.]+ |
segment SEQuence |
|
11 |
QUAL |
String |
[!-~]+ |
ASCII of Phred-scaled base QUALity+33 |
FLAG는 read가 reference에 어떤 방식으로 mapping 되었는지 숫자로 표기한 항목입니다. 각 숫자가 의미하는 바는 아래와 같습니다.

Primary alignment는 가장 높은 score를 가진 alignment를 의미합니다. 즉, 하나의 read는 reference의 여러 부위에 붙을 수 있는데 단 하나의 alignment만 primary가 되고 그 외 나머지는 모두 secondary가 됩니다. Secondary alignment는 downstream analysis에서 보통 사용되지 않습니다.
Supplementary alignment를 이해하기 위해서 우선 linear alignment를 알아야 하는데, 이는 하나의 read가 방향변화 없이 reference에 alignment 된 것을 의미합니다. Linear alignment가 아닌 read를 모두 chimeric alignment라고 합니다. 통상적으로 chimeric alignments 중 하나의 linear alignment를 representative alignment라고 하며 그 외 나머지를 supplementary alignment라고 합니다.
CIGAR string은 read의 sequence가 reference에 mapping된 양상을 문자와 숫자로 표기한 항목입니다. Read에서 몇 개의 base가 align (match or mismatch) 되었는지, 사라졌는지 (deletion), 추가되었는지 (insertion) 등의 정보를 확인할 수 있습니다. 각 CIGAR string의 문자가 의미하는 바는 아래와 같습니다.

그 외 SAM 파일 관련 정보는 SAM Format Specification을 참고하세요.
SAM 파일의 용량을 줄이기 위해 보통 BAM binary alignment map 파일 형태로 변환한 뒤 이후 분석에 사용합니다. BAM 파일 변환은 'samtools' 프로그램을 사용합니다.
BAM QC
BAM 파일 QC 단계입니다.
Human reference에 전체 시퀀싱 reads 중 어느 정도 mapping이 되었는지, duplicate reads의 비율은 어느 정도인지, concordant reads 비율은 어느 정도인지 등을 확인한 뒤 분석 목적에 따라 적절한 방안을 마련해야 올바른 variant를 찾을 수 있습니다.
'samtools stats'은 BAM 파일에 대한 전반적인 통계치를 생성합니다. 결과값 각 행은 CHK Checksum, SN Summary numbers, FFQ First fragment qualities 등의 tag로 시작하는데, 주요 내용은 SN tag에 정리되어 있으며 시퀀싱 reads, mapping 결과를 수치로 확인할 수 있습니다.

'plot-bamstats'를 사용하여 위 결과를 그래프로 표현할 수 있습니다. 마찬가지로 samtools의 기능 중 하나이며 수치로 나타난 결과를 한 눈에 보기 좋은 그래프로 표현해 줍니다.


'samtools idxstats'은 indexing된 BAM 파일의 index 정보를 바탕으로 통계치를 생성합니다. Human reference를 사용한 경우 각 chromosome별 name / length / #mapped-read segments / #unmapped read-segments 정보를 출력합니다.

이외에도 'picard', 'qualimap' 등의 프로그램을 사용해서 BAM QC 진행이 가능합니다.
Mark Duplicates
*** Warning: Amplicon sequencing, 혹은 동일한 start-end position을 target으로 하는 디자인을 사용한다면 본 과정을 수행하지 않습니다.
Duplicate를 처음 접해보신 분들은 생소한 개념일 것 입니다.
'똑같은, 꼭 닮은, 사본의' 라는 단어 자체의 의미와 같이 하나의 동일한 DNA fragment로부터 시퀀싱 된 read를 의미합니다. Clinical Data Overview (1/2) - 라이브러리 제작, 정제, QC에서 살펴본 바와 같이 DNA를 시퀀싱 하기 적당한 길이에 맞춰 무작위로 잘라낸 조각을 fragment라고 합니다. 수 백만 쌍의 genome을 무작위로 잘랐는데 그 fragment가 우연찮게 동일한 서열을 가진 같은 길이의 조각일 확률은 거의 없습니다. Repeat 영역으로 인해 실재하는 duplicate가 나올 수 있는 확률은 매우 미미한 수준일 것 입니다.
하지만 실제 시퀀싱 data를 살펴보면 생각보다 많은 duplicate를 발견할 수 있습니다. Illumina 플랫폼에서는 아래와 같은 원인에 의해 duplicate가 생성됩니다.

Optical duplicate: 시퀀싱을 진행하면서 cluster로부터 나오는 signal을 RTA software가 해석하는데, 이 때 하나의 커다란 cluster에서 나오는 signal을 두 개의 분리된 signal로 해석하면서 생기는 duplicate입니다. Non-patterned flowcell을 사용하는 HiSeq 2500, MiSeq, NextSeq 등에서 나타나는 현상입니다.
Clustering duplicate: 반대로 Patterned flowcell을 사용하는 HiSeq 4000, HiSeq X, NovaSeq 등에서 나타나는 현상입니다. 각 flowcell은 한 개의 library가 cluster를 형성해야 하는데, bridge amplification 과정에서 library가 인접한 flowcell까지 cluster를 형성한 경우 생기는 duplicate입니다.
PCR duplicate: Flowcell은 표면에 5', 3' adapter oligo가 부착되어 있습니다. Library 생성 후 flowcell에 로딩하면 서로 상보적인 서열이 결합하는 화학반응이 일어납니다. 이 반응의 효율성은 곧 투입되는 library 양에 따라 달라지므로 PCR amplification을 통해 적정한 양으로 증폭시킵니다. 여기서 PCR을 몇 cycle 증폭시켜야 하는지에 대한 결정은 결국 library complexity에 달려있으며 샘플 특성에 알맞은 값을 찾아야 합니다. 이렇게 PCR 과정에서 생성되는 duplicate를 의미합니다.
Sister duplicate: dsDNA에서 양 가닥의 DNA strand가 서로 다른 cluster를 형성했을 때 나타나는 duplicate입니다.
Base Quality Score Recalibration
줄여서 BQSR로 표현합니다. 간단히 요약하면 시퀀서는 각각 called base마다 정확도(base quality score)를 예측하는데 이 때 발생하는 systematic errors를 보정하는 과정입니다.
앞서 phred score를 살펴 보았습니다. Q20은 99% 정확도를 의미하며 100개의 base call 결과 중 확률적으로 1개의 잘못된 결과가 나올 수 있음을 의미합니다. 꽤 괜찮은 수준으로 보이죠? 하지만 called base가 증가할수록 잘못된 결과도 증가합니다. 사람의 genome을 예로 들어볼까요? 30억(3 9)개의 서열로 이루어진 사람의 유전체를 30x로 시퀀싱 하면 900억(9 10)개의 서열을 얻을 수 있습니다. Q20 기준으로 잘못된 서열은 무려 9억(9 8)개나 됩니다. 따라서 잘못된 base quality score를 바로잡는 과정은 매우 중요합니다.
BQSR은 주요 두 단계를 거쳐 진행합니다. Input data (bam)과 known variants set을 기반으로 error model을 생성하는 단계 (BaseRecalibrator tool), 그리고 이 모델을 사용하여 base quality score를 보정한 새로운 bam 파일을 만드는 단계 (ApplyBQSR tool)입니다.
BaseRecalibrator와 ApplyBQSR은 모두 gatk에 포함된 tool 입니다.
Variant Calling
BQSR을 거친 bam 파일로부터 variant를 찾아내는 과정입니다.
Variant는 어디에서부터 기인했느냐에 따라 크게 germline과 somatic으로 분류됩니다. 사람은 아빠와 엄마의 생식세포가 만나서 수정된 후 발생과정을 거쳐 하나의 개체로 성장합니다. 이 때 생식세포(gamete)가 지니고 있는 variant는 분열하는 모든 세포에게 전달되며 (ingeritance) 발생한 개체의 모든 세포 또한 동일한 variant를 공유하게 됩니다. (Mosaicism) 이런 variant가 germline 입니다.
반면 발생과정 동안 생긴 variant나 자외선, 흡연 등의 위험요소에 노출되어 생긴 variant는 일부 체세포에만 존재하며 후대에 전달되지 않습니다. 이런 variant가 somatic 입니다.

Breast/hereditary cancer와 같은 유전성 cancer는 germline variant를 검출합니다. 그 외 대부분 cancer는 somatic variant로 인해 발생하므로 이를 검출합니다.
Variant Annotation
검출된 variant가 가지는 생물학적 의미와 인종 안에서 상대빈도를 DB로부터 찾아 연결시키는 과정을 annotation이라 합니다.
먼저 DB 종류 기준으로 살펴보면, 정상인이 가진 variant를 모아둔 population DB가 있습니다. 보통 건강에 이상이 없는 정상인이 WES 데이터로부터 검출된 variant의 frequency를 측정합니다.
dbSNP은 각 연구자가 검출한 human variant 및 이와 연관된 다양한 정보를 NCBI에 등록한 DB입니다. 각 variant는 rs number가 부여됩니다.
ExAC는 정상인 60,760명의 WES 자룔를 통합 분석하여 다양한 인종(라틴계, 동아시아인, 핀란드인, 유럽인, 동남아시아인, 아프리카인)에서 나타나는 variant별 빈도 등을 제공합니다. gnomAD는 125,748명의 WES 뿐만 아니라 15,708명의 WGS 자료를 통합 분석하여 다양한 인종에서 각 variant별 빈도 등을 제공합니다. ExAC과는 달리 INTRON 및 INTERGENIC 영역의 variant 정도 또한 제공합니다. 2021년 3월 기준 두 개 DB는 모두 gnomAD 한 곳의 사이트에 통합되어 제공되고 있습니다.
1000 Genomes Project는 26개 인구집단에서 선별된 2,504명에 대한 variant 정보를 제공합니다.
다음은 질환 돌연변이 관련 DB입니다. 특정 질환을 보이는 환자에게서 나타나는 variant를 모아 놓은 DB입니다. 유전설질환과 germline mutation 위주로 구성된 ClinVar, HGMD, LOVD 등이 있습니다. 또한 cancer mutation 위주의 COSMIC, TCGA, ICGC 등이 있습니다.
ClinVar는 의학적으로 중요한 variant와 genotype을 모아놓은 DB입니다. 연구 논문에서 발표된 variant와 임상 검사실에서 실제 환자의 유전검사를 판독하고 판정한 variant도 모여있습니다. NCBI에서 관리하는 DB로 업데이트 주기가 일주일 밖에 되지 않을 정도로 활발하게 논의가 이루어지고 있습니다. 검출된 variant의 clinical significance 정보를 확인할 때 가장 많이 활용하는 DB이기도 합니다.
HGMD는 인간 유전질환의 원인이 되거나 이와 관련 있는 germline mutation을 망라하여 정리한 DB입니다.
LOVD는 네덜란드에서 운영하는 DB로 genotype과 연관된 유전자 mutation에 대한 정보를 제공합니다.
COSMIC은 인간 cancer에서 발견되는 somatic mutation과 그 효과를 제공하는 DB입니다. SNP, indels, fusion, cnv에 대한 정보를 제공합니다.
TCGA는 미국 국립암연구소와 게놈연구소에서 주도적으로 진행한 프로젝트로서 30여개 암종에 대한 30,000 케이스 이상의 암 조직의 유전체 검사 결과를 모아 놓은 DB를 운용하고 있습니다.
ICGC는 TCGA를 포함한 다양한 암 유전체 연구 프로젝트에서 생성된 결과를 모아놓은 DB입니다.
Variant annotation은 SnpEff, annovar, VEP 프로그램을 주로 사용합니다. 해당 프로그램들은 variant에 DB 정보를 붙여줄 뿐만 아니라 variant로 인해 발생하는 amino acid의 변화까지 알려줍니다.
Interpretation and Reporting
Variant의 annotation 정보와 다양한 근거를 바탕으로 환자의 질환과 연관있는 variant를 찾아내고 해석하며 보고하는 NGS 검사의 마지막 단계입니다.
여러 가지 의학적 근거와 환자의 임상정보를 바탕으로 variant가 가진 임상적 중요성을 해석하고 판단해야 하므로 가장 중요하면서 매우 어려운 단계입니다.
Germline variant는 2015년 미국의학유전학회(The American College of Medical Genetics and Genomics, ACMG)에서 발간한 ACMG guideline을 variant interpretation 기준으로 사용합니다. 근거의 중요도와 개수에 따라 variant를 5단계로 분류하여 판정합니다. Pathogenic / Likely pathogenic / Uncertain significance / Likely Benign / Benign

Somatic variant는 2017년 분자병리학회(Association for Molecular Pathology, AMP)에서 발간한 AMP guideline을 variant interpretation 기준으로 사용합니다. 근거의 중요도와 기준에 따라 variant를 4개 tier로 분류하여 판정합니다. Tier I, Tier II, Tier III, Tier IV

데이터 분석 단계를 마치며
수고하셨습니다. 여러분은 암 환자 유전체 분석을 위한 데이터 생산 단계에 이어 분석 단계까지 살펴봤습니다. 특히 cancer 환자는 somatic variant calling이 분석의 주요 목표입니다. 결과를 토대로 환자의 상태를 진단하고 치료하는 보조 도구로 사용합니다. 더 나아가 신약개발, 바이오마커 발견 등 다양한 분야에 응용할 수 있는 genomic 데이터입니다. 최근에는 좀 더 정확하고 분석시간을 단축하기 위해서 머신러닝을 접목한 다양한 연구방법들이 발표되고 있습니다. 데이터 분석 뿐만이 아니라 머신러닝 활용 능력을 갖추는 것이 필수적인 시대가 다가오고 있습니다.
Cancer 환자의 DNA 데이터 분석과정을 살펴봤습니다. 이 글이 여러분에게 조금이나마 도움이 되기를 바랍니다.